
The first major reduction in AIDS-related mortality came with the introduction of protease inhibitors (PIs) as part of combination therapy during the mid- and late 1990s.1,2 Despite their significant role in slowing HIV progression, PIs—most notably, the first-generation compounds—have been associated with significant acute and chronic toxicities, including gastrointestinal disturbances; lipid, glucose, and metabolic abnormalities; and increased cardiovascular risk.3 Since then, second-generation PIs such as atazanavir and darunavir (DRV) have been associated with improved toxicity profiles, lower discontinuation rates, and increased patient convenience.4,5
Darunavir was first approved by the FDA in 2006, and has a long track record of efficacy and safety in treatment-naive and -experienced patients within and outside the clinical trial setting.5-9 Current US guidelines for antiretroviral use do not recommend PI-based regimens for initial treatment for most adults and adolescents with HIV. However, DRV-based therapy is often considered an alternative in specific cases, such as when treatment initiation cannot be delayed until resistance test results are available, during pregnancy, and in cases of resistant HIV-1.10,11 Additionally, DRV-based therapy is also recommended in certain pediatric populations.12 Finally, owing to a low prevalence of resistance in the community, a high barrier to resistance, and low emergence of treatment-related resistance, DRV may be appropriate when the development of HIV-1 resistance is a concern.10
Tenofovir (TFV), as part of a dual nucleoside/nucleotide reverse transcriptase inhibitor (NRTI) backbone, can be coadministered with emtricitabine (FTC) in coformulated products, either as tenofovir disoproxil fumarate (TDF) or tenofovir alafenamide (TAF).10 The newer version (TAF) has been associated with lower renal and bone toxicity compared with the older formulation (TDF), particularly as part of boosted regimens containing ritonavir or cobicistat (COBI).13-15 DRV/COBI/FTC/TAF (Symtuza, Janssen), approved in July 2018, is the first single-tablet, PI-based regimen for the treatment of HIV-1 in antiretroviral-naive and -experienced adults, combining DRV, COBI (a pharmacokinetic enhancer or “booster”), FTC, and TAF in a single product (Table 1).16

| Table 1. Clinical Pearls of the Coformulated STR of DRV/COBI/FTC/TAF (Symtuza, Janssen) |
| Once-daily regimen, which is associated with improved adherence10,20-22 |
| STR adds convenience, ease of use, and can improve adherence23-27 |
| Contains DRV, which has a high genetic barrier to resistance30 |
| Each individual component has a long track of efficacy and safety5-10,13-15 |
| As a single-tablet combination, demonstrated high efficacy, low toxicity, and tolerability17-19 |
| DRV/COBI/FTC/TAF, darunavir-cobicistat-emtricitabine-tenofovir alafenamide; STR, single-tablet regimen |
Clinical Efficacy and Safety
A phase 2, randomized, double-blind, multicenter, active-controlled study evaluated the efficacy and safety of a DRV 800-mg/COBI 150-mg/FTC 200-mg/TAF 10-mg single-tablet regimen (STR; D/C/F/TAF) in 103 patients compared with a 2-DRV 400-mg plus COBI 150-mg plus FTC 200-mg/TDF 300-mg FTC 200-mg/TDF 300-multiple-tablet regimen (MTR; D+C+F/TDF; Truvada, Gilead) in 50 patients; each arm included matching placebo.17 The study population included treatment-naive, HIV-positive people with virus susceptible to DRV, FTC, and TFV, and an estimated glomerular filtration rate (eGFR) of at least 70 mL per minute. Patients were excluded if they were pregnant, had hepatitis B or C virus (HBV, HCV), or had a new AIDS-defining illness within 30 days of the screening visit. In addition to routine safety and efficacy evaluations, subsets of patients underwent sampling for population pharmacokinetics and determination of intracellular TFV diphosphate concentrations in peripheral blood mononuclear cells. The primary objective was the efficacy of D/C/F/TAF versus D+C+F/TDF, defined as HIV-1 RNA less than 50 copies/mL at week 24 in the intent-to-treat snapshot algorithm, per the FDA.17 Secondary end points included undetectable HIV-1 RNA at week 48 and efficacy in selected subgroups.
Baseline characteristics were similar between arms, with a median age of 33 years (range, 26-43 years), 92.8% men, 34.6% black patients, 21% Hispanic patients, 19.6% with an HIV-1 RNA greater than 100,000 copies/mL, 13.7% with a CD4+ T-cell count less than 200 cells/mm3, and a median eGFR of 114.6 mL per minute (range, 96.5-132.3 mL/min). The primary end point of undetectable HIV-1 RNA at week 24 was observed in 74.8% on D/C/F/TAF versus 74.0% on D+C+F/TDF, with a weighted difference of 3.3% (95% CI, 211.4%-18.1%). This difference fell within the prespecified no-greater-than –12% margin, although this study was not powered for establishing noninferiority. At week 48, undetectable HIV-1 RNA was observed in 76.7% on D/C/F/TAF versus 84.0% on D+C+F/TDF (weighted difference, –6.2%; 95% CI, –19.9 to 7.4), which was driven primarily by reasons other than virologic failure (lost to follow-up/investigator decision). Subgroup analysis did not observe significant differences in efficacy between arms. Of note, virologic failure at weeks 24 and 48, respectively, occurred in 20% and 16% of STR patients versus 24% and 12% of MTR patients, although none of these failures were attributed to development of viral resistance.
Treatment-emergent adverse events (TEAEs) were observed in 92.2% (95/103) of STR patients versus 94.0% (47/50) of MTR patients, most of which were mild or moderate in severity. The following TEAEs were reported in at least 10% of patients taking D/C/F/TAF versus D+C+F/TDF: diarrhea, 21.4% versus 26%; upper respiratory tract infection, 15.5% versus 14%; fatigue, 13.6% versus 18%; nausea, 12.6% versus 10%; rash, 11.7% versus 8%; flatulence, 4.5% versus 12%; pain in extremity, 7.8% versus 10%; vitamin D deficiency, 1.9% versus 10%; and vomiting, 3.9% versus 10%. Serum creatinine in the D/C/F/TAF arm compared with D+C+F/TDF increased from baseline to week 48 by 0.06 mg/dL (95% CI, 0.04-0.08 mg/dL) versus 0.09 mg/dL (95% CI, 0.05-0.14 mg/dL; P=0.053).17 The median decrease in eGFR from baseline to week 48 was –2.9 mL per minute for the D/C/F/TAF group versus –10.6 mL per minute for the D+C+F/TDF group (P=0.017). The median changes in lipids from baseline to week 48 for the STR versus MTR groups were 40 versus 5 mg/dL in total cholesterol (P<0.001); 26 versus 4 mg/dL in low-density lipoprotein (P<0.001); 7 versus 3 mg/dL in high-density lipoprotein (P=0.009); and 29 versus –5 mg/dL for triglycerides (P=0.007). Overall, the comparable efficacy and favorable toxicity of the STR versus MTR groups supported the progression of D/C/F/TAF to larger phase 3 trials in treatment-naive and -experienced adults with HIV-1.
The first of these studies, known as the AMBER trial, was a phase 3, randomized, double-blind, multicenter, active-controlled trial designed to evaluate the efficacy and safety of D/C/F/TAF (n=362) versus D+C+F/TDF (n=363) in HIV-1-infected, treatment-naive adults.18 This noninferiority study included adults with HIV-1 RNA less than 1,000 copies/mL, a CD4+ T-cell count greater than 200 cells/mm3, susceptibility to all active study medications, without HBV or HCV coinfection, and who were not pregnant. The primary end point was the proportion of virologic suppression (HIV-1 RNA <50 copies/mL) through week 48 by the FDA snapshot analysis, with the lower limit of the 2-sided 95% CI of the difference (study minus control) greater than –10 percentage points as noninferiority criteria. Secondary outcomes included undetectability by different limits of HIV-1 RNA detection and by different baseline or disease severity characteristics, changes from baseline in viral load and CD4+ T-cell count, de novo resistance, and safety and tolerability.
Participants were from North America and Europe; the median age was 34 years (range, 27-42 years); 88% were men; 83% were white; 13% were Hispanic or Latino; 7% had a CD4+ T-cell count less than 200 cells/mm3 (median, 453 cells/mm3; range, 333-601 cells/mm3); 18% had a viral load of at least 100,000 copies/mL; and the overall median eGFR was 119.1 mL per minute (range, 104.4-136.5 mL/min).18 For the primary outcome at week 48, 91.4% (331/362) of participants on D/C/F/TAF versus 88.4% (321/363) on D+C+F/TDF achieved HIV-1 RNA less than 50 copies/mL (difference of 2.7 percentage points; 95% CI, –1.6 to 7.1; P<0.0001). Noninferiority also was confirmed with respect to baseline characteristics and disease severity, except for a CD4+ T-cell count less than 200 cells/mm3, black race, and age older than 50 years (Table 2).
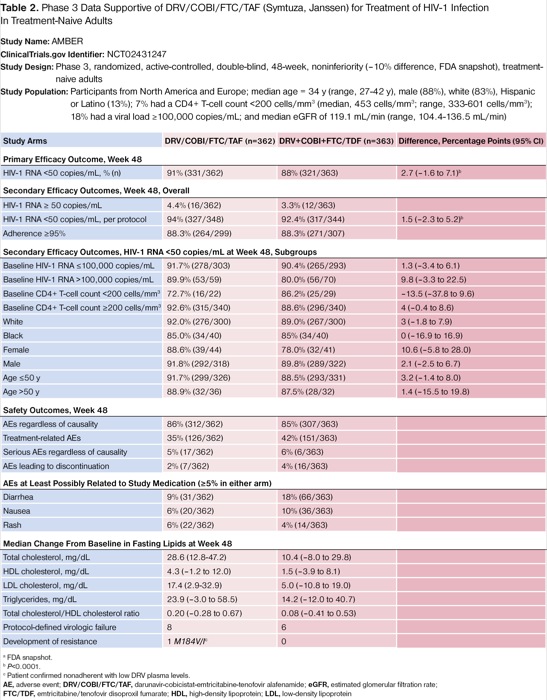
| Table 2. Phase 3 Data Supportive of DRV/COBI/FTC/TAF (Symtuza, Janssen) for Treatment of HIV-1 Infection In Treatment-Naive Adults Study Name: AMBER ClinicalTrials.gov Identifier: NCT02431247 Study Design: Phase 3, randomized, active-controlled, double-blind, 48-week, noninferiority (–10% difference, FDA snapshot), treatment-naive adults Study Population: Participants from North America and Europe; median age = 34 y (range, 27-42 y), male (88%), white (83%), Hispanic or Latino (13%); 7% had a CD4+ T-cell count <200 cells/mm3 (median, 453 cells/mm3; range, 333-601 cells/mm3); 18% had a viral load ≥100,000 copies/mL; and median eGFR of 119.1 mL/min (range, 104.4-136.5 mL/min) | |||
| Study Arms | DRV/COBI/FTC/TAF (n=362) | DRV+COBI+FTC/TDF (n=363) | Difference, Percentage Points (95% CI) |
|---|---|---|---|
| Primary Efficacy Outcome, Week 48 | |||
| HIV-1 RNA <50 copies/mL, % (n) | 91% (331/362) | 88% (321/363) | 2.7 (–1.6 to 7.1)b |
| Secondary Efficacy Outcomes, Week 48, Overall | |||
| HIV-1 RNA ≥ 50 copies/mL | 4.4% (16/362) | 3.3% (12/363) | |
| HIV-1 RNA <50 copies/mL, per protocol | 94% (327/348) | 92.4% (317/344) | 1.5 (–2.3 to 5.2)b |
| Adherence ≥95% | 88.3% (264/299) | 88.3% (271/307) | |
| Secondary Efficacy Outcomes, HIV-1 RNA <50 copies/mL at Week 48, Subgroups | |||
| Baseline HIV-1 RNA ≤100,000 copies/mL | 91.7% (278/303) | 90.4% (265/293) | 1.3 (–3.4 to 6.1) |
| Baseline HIV-1 RNA >100,000 copies/mL | 89.9% (53/59) | 80.0% (56/70) | 9.8 (–3.3 to 22.5) |
| Baseline CD4+ T-cell count <200 cells/mm3 | 72.7% (16/22) | 86.2% (25/29) | –13.5 (–37.8 to 9.6) |
| Baseline CD4+ T-cell count ≥200 cells/mm3 | 92.6% (315/340) | 88.6% (296/340) | 4 (–0.4 to 8.6) |
| White | 92.0% (276/300) | 89.0% (267/300) | 3 (–1.8 to 7.9) |
| Black | 85.0% (34/40) | 85% (34/40) | 0 (–16.9 to 16.9) |
| Female | 88.6% (39/44) | 78.0% (32/41) | 10.6 (–5.8 to 28.0) |
| Male | 91.8% (292/318) | 89.8% (289/322) | 2.1 (–2.5 to 6.7) |
| Age ≤50 y | 91.7% (299/326) | 88.5% (293/331) | 3.2 (–1.4 to 8.0) |
| Age >50 y | 88.9% (32/36) | 87.5% (28/32) | 1.4 (–15.5 to 19.8) |
| Safety Outcomes, Week 48 | |||
| AEs regardless of causality | 86% (312/362) | 85% (307/363) | |
| Treatment-related AEs | 35% (126/362) | 42% (151/363) | |
| Serious AEs regardless of causality | 5% (17/362) | 6% (6/363) | |
| AEs leading to discontinuation | 2% (7/362) | 4% (16/363) | |
| AEs at Least Possibly Related to Study Medication (≥5% in either arm) | |||
| Diarrhea | 9% (31/362) | 18% (66/363) | |
| Nausea | 6% (20/362) | 10% (36/363) | |
| Rash | 6% (22/362) | 4% (14/363) | |
| Median Change From Baseline in Fasting Lipids at Week 48 | |||
| Total cholesterol, mg/dL | 28.6 (12.8-47.2) | 10.4 (–8.0 to 29.8) | |
| HDL cholesterol, mg/dL | 4.3 (–1.2 to 12.0) | 1.5 (–3.9 to 8.1) | |
| LDL cholesterol, mg/dL | 17.4 (2.9-32.9) | 5.0 (–10.8 to 19.0) | |
| Triglycerides, mg/dL | 23.9 (–3.0 to 58.5) | 14.2 (–12.0 to 40.7) | |
| Total cholesterol/HDL cholesterol ratio | 0.20 (–0.28 to 0.67) | 0.08 (–0.41 to 0.53) | |
| Protocol-defined virologic failure | 8 | 6 | |
| Development of resistance | 1 M184V/Ic | 0 | |
a FDA snapshot. b P<0.0001. c Patient confirmed nonadherent with low DRV plasma levels. AE, adverse event; DRV/COBI/FTC/TAF, darunavir-cobicistat-emtricitabine-tenofovir alafenamide; eGFR, estimated glomerular filtration rate; FTC/TDF, emtricitabine/tenofovir disoproxil fumarate; HDL, high-density lipoprotein; LDL, low-density lipoprotein | |||
The safety profiles of D/C/F/TAF and D+C+F/TDF were similar in the AMBER trial (Table 2).18 Although TEAEs, including gastrointestinal AEs, were numerically higher for the D+C+F/TDF regimen, the lipid-protective effects of TDF regarding non–high-density lipoprotein cholesterol were recognizable in this study arm. There were 8 patients in the D/C/F/TAF group and 6 in the D+C+F/TDF arm with protocol-defined virologic failure by week 48. An M184V/I mutation against FTC was identified in a patient in the D/C/F/TAF arm who also had a baseline non-NRTI mutation (K103N). Low DRV plasma concentrations indicated likely nonadherence to treatment. No resistance mutations against DRV, TDF, or TAF were observed by week 48.
{RELATED-VERTICAL}A second phase 3, randomized, double-blind, multicenter, active-controlled study, known as the EMERALD trial, assessed the noninferiority of switching to D/C/F/TAF (n=763) versus continuing a boosted PI (bPI) plus F/TDF (n=378) in virologically suppressed, HIV-1-infected adults already on a bPI-based regimen.19 This noninferiority study included patients with HIV-1 RNA less than 50 copies/mL within 2 months of screening, without resistance mutations against DRV, negative for HBV or HCV coinfection, and who were not pregnant. Additionally, participants had to be stable on their bPI plus F/TDF regimen for at least 6 months, and the bPI could include ritonavir- or COBI-boosted DRV or atazanavir, or ritonavir-boosted lopinavir. The primary objective of noninferiority between the D/C/F/TAF (STR) and bPI plus F/TDF (MTR) regimens was assessed as the proportion of virologic rebound (HIV-1 RNA ≥50 copies/mL) through week 48, with the upper limit of the 2-sided 95% CI of the difference (study minus control) less than 4 percentage points as noninferiority criteria. Secondary outcomes included undetectability by different limits of HIV-1 RNA detection and by different baseline or disease severity characteristics, changes from baseline in viral load and CD4+ T-cell count, de novo resistance, and safety and tolerability.
Baseline characteristics were again similar between groups in the EMERALD and the AMBER trials. Participants were from North America and Europe; the median age was 46 years (range, 19-78 years); 82% were men; 75% were white; 15% were Hispanic or Latino; the median CD4+ T-cell count was less than 200 cells/mm3 (median, 628 cells/mm3; range, 468-802 cells/mm3); 70% were using boosted DRV; 22% were using boosted atazanavir; and 8% were using boosted lopinavir.19 For the primary end point at week 48, 2.5% (19/763) of participants on D/C/F/TAF versus 2.1% (8/378) on D+C+F/TDF had rebound HIV-1 RNA of 50 copies/mL or more (difference of 0.4 percentage points; 95% CI, –1.5 to 2.2; P<0.0001). A per-protocol analysis showed consistent results, where 1.9% (14/721) versus 0.8% (3/358) had rebound HIV-1 RNA of at least 50 copies/mL (difference of 1.1 percentage points; 95% CI, –0.3 to 2.5; P<0.0001). Stratified analyses demonstrated noninferiority with respect to baseline characteristics and antiretroviral history (Table 3).
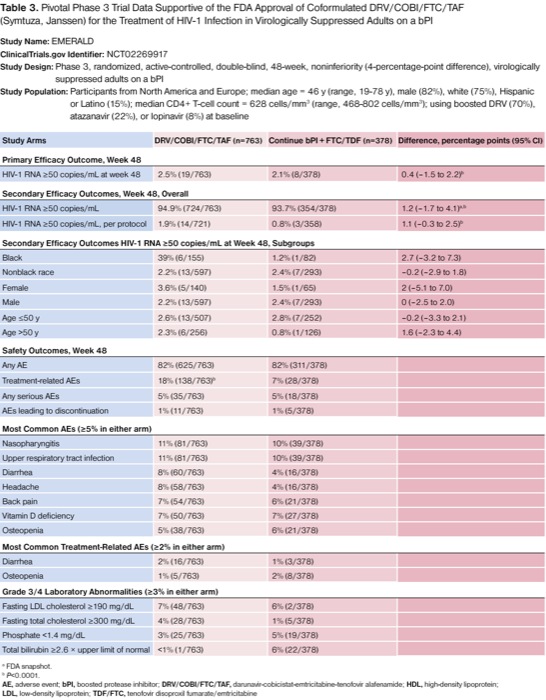
| Table 3. Pivotal Phase 3 Trial Data Supportive of the FDA Approval of Coformulated DRV/COBI/FTC/TAF (Symtuza, Janssen) for the Treatment of HIV-1 Infection in Virologically Suppressed Adults on a bPI Study Name: EMERALD ClinicalTrials.gov Identifier: NCT02269917 Study Design: Phase 3, randomized, active-controlled, double-blind, 48-week, noninferiority (4-percentage-point difference), virologically suppressed adults on a bPI Study Population: Participants from North America and Europe; median age = 46 y (range, 19-78 y), male (82%), white (75%), Hispanic or Latino (15%); median CD4+ T-cell count = 628 cells/mm3 (range, 468-802 cells/mm3); using boosted DRV (70%), atazanavir (22%), or lopinavir (8%) at baseline | |||
| Study Arms | DRV/COBI/FTC/TAF (n=763) | Continue bPI + FTC/TDF (n=378) | Difference, percentage points (95% CI) |
|---|---|---|---|
| Primary Efficacy Outcome, Week 48 | |||
| HIV-1 RNA ≥50 copies/mL at week 48 | 2.5% (19/763) | 2.1% (8/378) | 0.4 (–1.5 to 2.2)b |
| Secondary Efficacy Outcomes, Week 48, Overall | |||
| HIV-1 RNA ≥50 copies/mL | 94.9% (724/763) | 93.7% (354/378) | 1.2 (–1.7 to 4.1)a,b |
| HIV-1 RNA ≥50 copies/mL, per protocol | 1.9% (14/721) | 0.8% (3/358) | 1.1 (–0.3 to 2.5)b |
| Secondary Efficacy Outcomes HIV-1 RNA ≥50 copies/mL at Week 48, Subgroups | |||
| Black | 39% (6/155) | 1.2% (1/82) | 2.7 (–3.2 to 7.3) |
| Nonblack race | 2.2% (13/597) | 2.4% (7/293) | –0.2 (–2.9 to 1.8) |
| Female | 3.6% (5/140) | 1.5% (1/65) | 2 (–5.1 to 7.0) |
| Male | 2.2% (13/597) | 2.4% (7/293) | 0 (–2.5 to 2.0) |
| Age ≤50 y | 2.6% (13/507) | 2.8% (7/252) | –0.2 (–3.3 to 2.1) |
| Age >50 y | 2.3% (6/256) | 0.8% (1/126) | 1.6 (–2.3 to 4.4) |
| Safety Outcomes, Week 48 | |||
| Any AE | 82% (625/763) | 82% (311/378) | |
| Treatment-related AEs | 18% (138/763)b | 7% (28/378) | |
| Any serious AEs | 5% (35/763) | 5% (18/378) | |
| AEs leading to discontinuation | 1% (11/763) | 1% (5/378) | |
| Most Common AEs (≥5% in either arm) | |||
| Nasopharyngitis | 11% (81/763) | 10% (39/378) | |
| Upper respiratory tract infection | 11% (81/763) | 10% (39/378) | |
| Diarrhea | 8% (60/763) | 4% (16/378) | |
| Headache | 8% (58/763) | 4% (16/378) | |
| Back pain | 7% (54/763) | 6% (21/378) | |
| Vitamin D deficiency | 7% (50/763) | 7% (27/378) | |
| Osteopenia | 5% (38/763) | 6% (21/378) | |
| Most Common Treatment-Related AEs (≥2% in either arm) | |||
| Diarrhea | 2% (16/763) | 1% (3/378) | |
| Osteopenia | 1% (5/763) | 2% (8/378) | |
| Grade 3/4 Laboratory Abnormalities (≥3% in either arm) | |||
| Fasting LDL cholesterol ≥190 mg/dL | 7% (48/763) | 6% (2/378) | |
| Fasting total cholesterol ≥300 mg/dL | 4% (28/763) | 1% (5/378) | |
| Phosphate <1.4 mg/dL | 3% (25/763) | 5% (19/378) | |
| Total bilirubin ≥2.6 × upper limit of normal | <1% (1/763) | 6% (22/378) | |
a FDA snapshot. b P<0.0001. AE, adverse event; bPI, boosted protease inhibitor; DRV/COBI/FTC/TAF, darunavir-cobicistat-emtricitabine-tenofovir alafenamide; HDL, high-density lipoprotein; LDL, low-density lipoprotein; TDF/FTC, tenofovir disoproxil fumarate/emtricitabine | |||
The safety profiles of D/C/F/TAF and bPI plus F/TDF were relatively similar in the EMERALD study (Table 3).19 Although any AEs, serious AEs, and AEs leading to discontinuation were similar across study arms, TEAEs were significantly higher for the D/C/F/TAF STR group versus the MTR group (18% vs. 7%; P<0.0001). Only 1 serious AE, pancreatitis, was deemed possibly related to D/C/F/TAF by the investigator. No resistance mutations against any PIs, FTC, TDF, or TAF were observed through week 48.
Place in Therapy
At the time of writing, 13 FDA-approved STRs are on the market in the United States,10 although DRV/COBI/FTC/TAF is the first STR consisting of a PI coformulated with an NRTI backbone. Among ideal antiretroviral regimen characteristics, once-daily dosing has been associated with improved treatment adherence compared with multiple-daily dosing.20-22 However, the impact of STRs versus MTRs is less defined. Studies comparing these two forms have observed improved adherence with STRs,23-27 but there are conflicting results with respect to clinical outcomes such as viral suppression, time to treatment discontinuation, failure, or hospitalization, among other end points.27-29 More rigorous randomized trials of STRs versus MTRs will likely be necessary to confirm the potential advantage of decreased pill burden.
There was minimal to no development of resistance during the registrational phase 2 and 3 studies for DRV/COBI/FTC/TAF. This is explained by DRV’s high genetic barrier to resistance, which is well characterized in the literature and has been observed in clinical practice.30 Antiretroviral regimens with a high genetic barrier to resistance, such as DRV/COBI/FTC/TAF, are recommended by US guidelines for individuals in whom poor adherence may be a concern to minimize the likelihood of emergence of resistance. Further, coformulation of individual antiretroviral components, each backed by years of DRV/COBI/FTC/TAF safety and efficacy data, provides treatment-naive and -experienced HIV-1-infected adult patients with another STR option that may add convenience and ease of use (Table 1).10
References
- Palella FJ Jr, et al. N Engl J Med. 1998;338(13):853-860.
- Gortmaker SL, et al. N Engl J Med. 2001;345(21):1522-1528.
- Margolis AM, et al. J Med Toxicol. 2014;10(1):26-39.
- Molina JM, et al. J Acquir Immune Defic Syndr. 2010;53(3):323-332.
- Orkin C, et al. HIV Med. 2013;14(1):49-59.
- FDA. bit.ly/2kq2ID6. Accessed August 5, 2019.
- Ortiz R, et al. AIDS. 2008;22(12):1389-1397.
- Molina JM, et al. Lancet HIV. 2015;2(4):e127-e136.
- Lennox JL, et al. Ann Intern Med. 2014;161(7):461-471.
- Panel on Antiretroviral Guidelines for Adults and Adolescents. bit.ly/2lQ9sum. Accessed August 5, 2019.
- Panel on Treatment of HIV-Infected Pregnant Women and Prevention of Perinatal Transmission. bit.ly/2kpZ503. Accessed August 5, 2019.
- Panel on Antiretroviral Therapy and Medical Management of Children Living With HIV. Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection. bit.ly/2jTgQEG. Accessed August 5, 2019.
- DeJesus E, et al. AIDS Res Hum Retroviruses. 2018;34(4):337-342.
- Mills A, et al. Lancet Infect Dis. 2016;16(1):43-52.
- Hill A, et al. J Vir Erad. 2018;4(2):72-79.
- FDA. bit.ly/2lu2zyX. Accessed August 5, 2019.
- Mills A, et al. J Acquir Immune Defic Syndr. 2015;69(4):439-445.
- Eron JJ, et al. AIDS. 2018;32(11):1431-1442.
- Orkin C, et al. Lancet HIV. 2018;5(1):e23-e34.
- Parienti JJ, et al. Clin Infect Dis. 2009;48(4):484-488.
- Raboud J, et al. AIDS Behav. 2011;15(7):1397-1409.
- Nachega JB, et al. Clin Infect Dis. 2014;58(9):1297-1307.
- Bangsberg DR, et al. AIDS. 2010;24(18):2835-2840.
- Clay PG, et al. Medicine (Baltimore). 2015;94(42):e1677.
- Sutton ss, et al. Am J Manag Care. 2016;22(4):242-248.
- Sutton ss, et al. Pharmacotherapy. 2016;36(4):383-401.
- Cotte L, et al. PLoS One. 2017;12(2):e0170661.
- Drozd DR, et al. Medicine (Baltimore). 2017;96(14):e6275.
- Hemmige V, et al. AIDS Care. 2018;30(8):1017-1024.
- Aoki M, et al. MBio. 2018;9(2):e02425-17.
About the authors:

